Annotation of 10x 8k PBMCs from a Healthy Donor dataset
Viktor Petukhov
2018-01-24
Source file: notebooks/annotation/annotation_pbmc8k.Rmd
Last updated: 2018-05-29
Code version: 3ea6d7c
Read data
holder <- readRDS(paste0(kEstFolder, 'pbmc8k_no_umi.rds'))
genes <- read.table(paste0(k10xFolder, 'genes.tsv')) %>%
filter(V2 %in% names(which(table(V2) == 1)))
gene_id_to_names <- setNames(genes$V2, genes$V1)
holder$cm_raw <- holder$cm_raw[grep("^[^;]+$", rownames(holder$cm_raw)),]umis_per_cell <- sort(Matrix::colSums(holder$cm_raw), decreasing=T)
est_cell_num <- EstimateCellsNumber(umis_per_cell)scores <- ScorePipelineCells(holder, mit.chromosome.name='MT',
predict.all=T)[names(umis_per_cell)]
PlotCellScores(scores, cells.number=est_cell_num)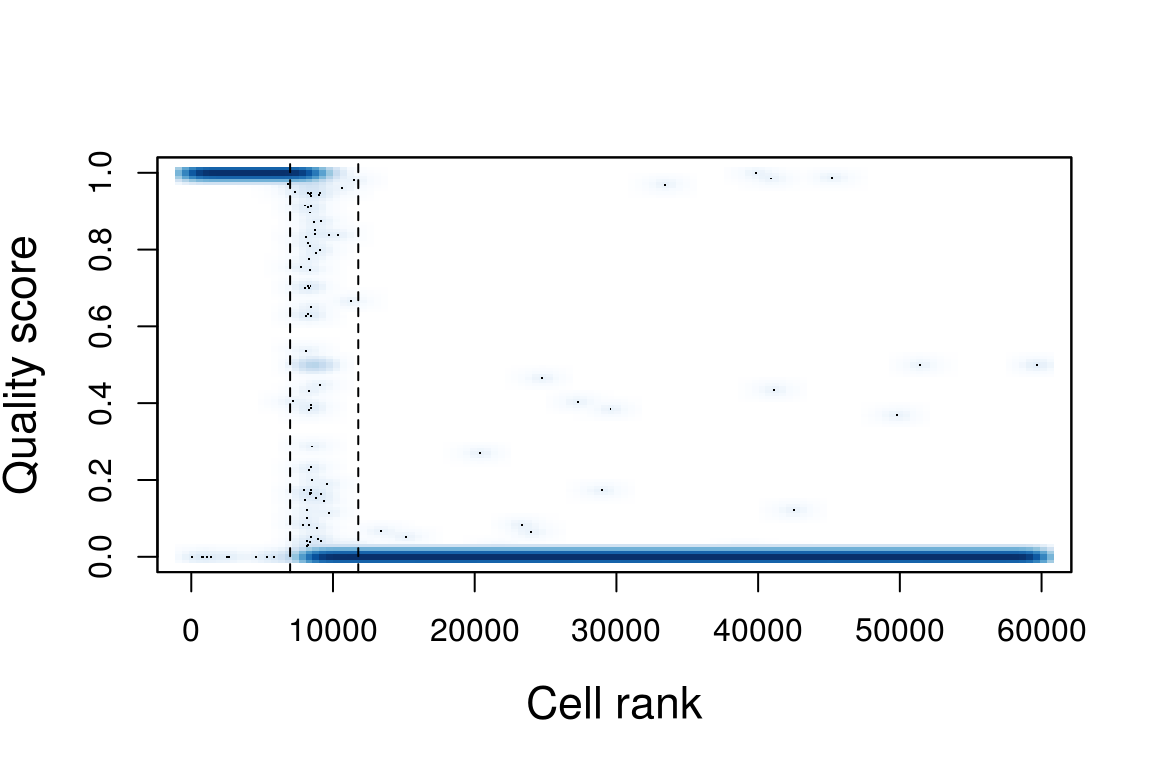
Pagoda run:
real_cbs <- names(scores)[1:est_cell_num$expected]
real_cbs <- real_cbs[scores[real_cbs] > 0.9]
r_cm <- holder$cm_raw[, real_cbs]
r_cm <- r_cm[intersect(rownames(r_cm), names(gene_id_to_names)), ]
rownames(r_cm) <- gene_id_to_names[rownames(r_cm)]
pgd <- GetPagoda(r_cm, n.cores=30)8451 cells, 12931 genes; normalizing ... using plain model winsorizing ... log scale ... done.
calculating variance fit ... using gam 485 overdispersed genes ... 485 persisting ... done.
running PCA using 1000 OD genes .... done
calculating distance ... pearson ...running tSNE using 30 cores:# clusters <- pgd$clusters$PCA$infomap
# write.csv(clusters, paste0(kAnnotationData, 'pbmc8k_clusters.csv'))
# Pagoda uses stochastic clustering algorithm, so we saved clusters from one run
clusters <- read.csv(paste0(kAnnotationData, 'pbmc8k_clusters.csv'), row.names=1)
clusters <- setNames(clusters$x, rownames(clusters))
log_mtx <- log10(1e-3 + as.matrix(pgd$counts[names(clusters), ]))Initial clustering:
PlotPagodaEmbeding(pgd, clusters=clusters, show.ticks=F)
Initial labeling
de_genes <- pgd$getDifferentialGenes(type='PCA', groups=clusters,
upregulated.only=T) %>% lapply(rownames)
major_cell_types <- lst(
`T cells` = sapply(de_genes, function(genes) 'CD3D' %in% genes) %>%
which() %>% names() %>% as.integer(),
`B cells` = sapply(de_genes, function(genes) 'MS4A1' %in% genes) %>%
which() %>% names() %>% as.integer()
)
major_type_clusts <- major_cell_types %>% unlist()
if (length(major_type_clusts) != length(unique(major_type_clusts)))
stop("Something goes wrong")heatmap_genes <- c(
'MS4A1',
'CD3D', 'CD3E',
'LYZ', 'CD14',
'GNLY', 'NKG7',
"GZMA", "GZMB", "GZMH", "GZMK",
'FCGR3A', 'MS4A7',
'FCER1A', 'CST3',
'PPBP')
heatmap_clusters <- clusters[!(clusters %in% unlist(major_cell_types))]
# heatmap_clusters <- heatmap_clusters[heatmap_clusters > 20]
PlotExpressionHeatmap(log_mtx, heatmap_clusters, heatmap_genes)
type_ids <- c(major_cell_types, lst(
`CD14+ Monocytes` = c(1, 12, 13, 34),
`NK cells 1` = c(9, 29, 30),
`NK cells 2` = 26,
`FCGR3A+ Monocytes` = 15,
`Dendritic cells` = c(18, 24),
`Megakaryocytes` = 31
))
type_ids$`T cells` <- c(type_ids$`T cells`, 35)
markers_df <- data.frame(
Type = c(
"B cells", "T cells", "CD14+ Monocytes", "NK cells 1", "NK cells 2",
"FCGR3A+ Monocytes", "Dendritic cells", "Megakaryocytes"),
Markers = c(
"MS4A1", "CD3D, CD3E", "LYZ, CD14", "GNLY, NKG7", "GZMB, NKG7",
"FCGR3A, MS4A7", "FCER1A, CST3", "PPBP")
)
markers_df$Clusters <- sapply(type_ids, paste, collapse=", ")[as.character(markers_df$Type)]
markers_df| Type | Markers | Clusters |
|---|---|---|
| B cells | MS4A1 | 4, 8, 19, 22, 23, 28, 29, 33 |
| T cells | CD3D, CD3E | 2, 3, 5, 6, 7, 10, 11, 14, 16, 17, 20, 21, 25, 27, 32, 35 |
| CD14+ Monocytes | LYZ, CD14 | 1, 12, 13, 34 |
| NK cells 1 | GNLY, NKG7 | 9, 29, 30 |
| NK cells 2 | GZMB, NKG7 | 26 |
| FCGR3A+ Monocytes | FCGR3A, MS4A7 | 15 |
| Dendritic cells | FCER1A, CST3 | 18, 24 |
| Megakaryocytes | PPBP | 31 |
clusters_annotated <- AnnotateClusters(clusters, type_ids)
PlotClustering(pgd, clusters_annotated)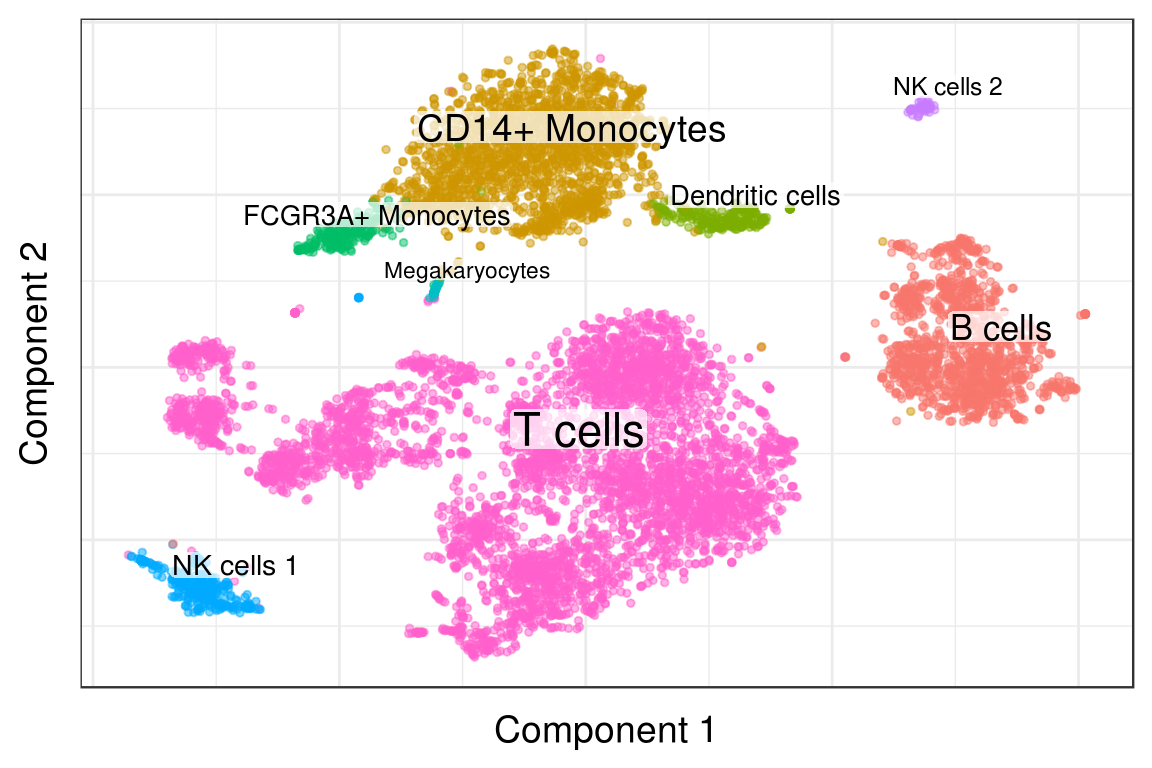
T Cells
heatmap_genes <- c(
"CD3D", "CD3E", "CCR7",
"CD8A", "CD8B",
"IL7R", "CD4",
"NKG7", "GZMA", "GZMH", "GZMK"
)
heatmap_clusters <- clusters[clusters %in% type_ids$`T cells`]
# heatmap_clusters <- heatmap_clusters[heatmap_clusters > 3]
PlotExpressionHeatmap(log_mtx, heatmap_clusters, heatmap_genes)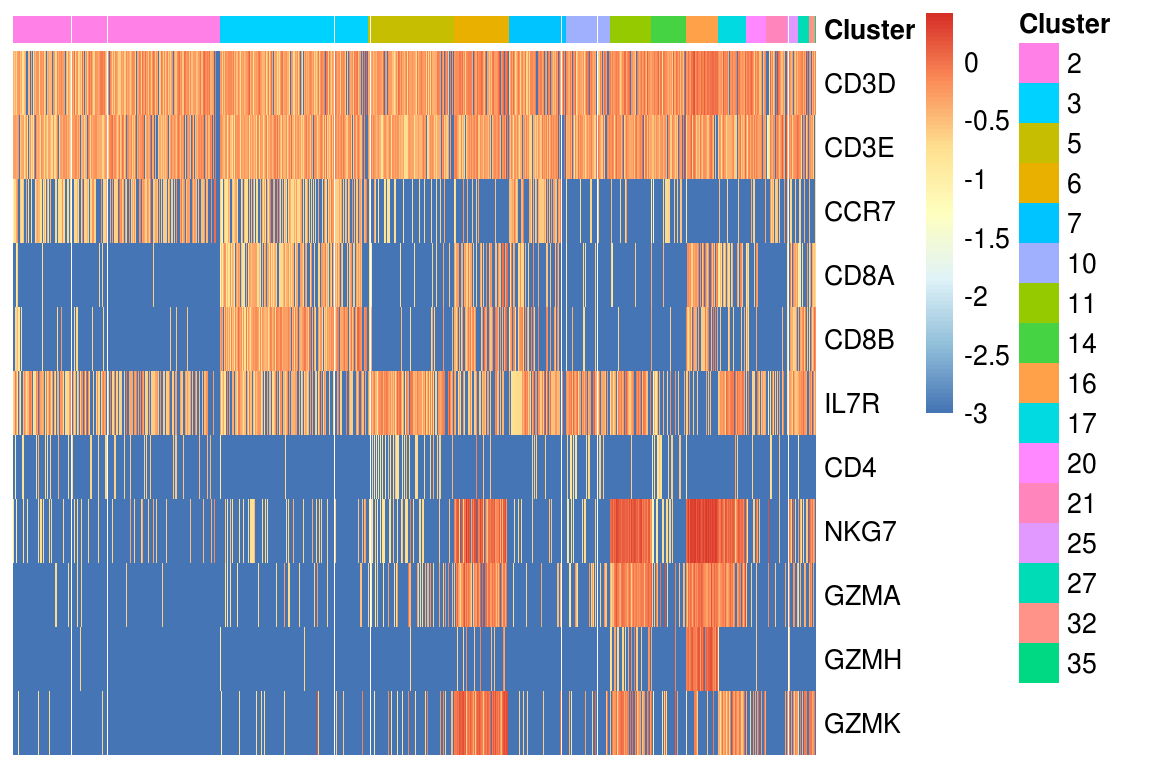
t_markers_df <- data.frame(
Type = c("Naive T cells", "CD8 T cells", "CD4+ T cells", "Cytotoxic T cells"),
Markers = c("CCR7", "CD8A, CD8B", "IL7R, CD4", "NKG7, GZMA, GZMH, GZMK")
)
type_ids <- c(type_ids, lst(
`Naive T cells` = c(2, 7, 21),
`CD8 T cells` = c(3),
`CD4+ T cells` = c(5, 10, 14, 35),
`Cytotoxic T cells` = c(6, 11, 16, 17, 20, 25, 27, 32)
))
type_ids$`T cells` <- NULL
t_markers_df$Clusters <- sapply(type_ids, paste, collapse=", ")[as.character(t_markers_df$Type)]
t_markers_df| Type | Markers | Clusters |
|---|---|---|
| Naive T cells | CCR7 | 2, 7, 21 |
| CD8 T cells | CD8A, CD8B | 3 |
| CD4+ T cells | IL7R, CD4 | 5, 10, 14, 35 |
| Cytotoxic T cells | NKG7, GZMA, GZMH, GZMK | 6, 11, 16, 17, 20, 25, 27, 32 |
clusters_annotated <- AnnotateClusters(clusters, type_ids)
write.csv(data.frame(Barcode=names(clusters_annotated),
Type=as.vector(clusters_annotated)),
paste0(kAnnotationData, 'pbmc8k_clusters_annotated.csv'))All markers
all_markers_df <- rbind(markers_df, t_markers_df)
write.csv(all_markers_df[c('Type', 'Markers')],
paste0(kOutputPath, 'tables/annotation_pbmc8k_markers.csv'), row.names=F)
all_markers_df| Type | Markers | Clusters |
|---|---|---|
| B cells | MS4A1 | 4, 8, 19, 22, 23, 28, 29, 33 |
| T cells | CD3D, CD3E | 2, 3, 5, 6, 7, 10, 11, 14, 16, 17, 20, 21, 25, 27, 32, 35 |
| CD14+ Monocytes | LYZ, CD14 | 1, 12, 13, 34 |
| NK cells 1 | GNLY, NKG7 | 9, 29, 30 |
| NK cells 2 | GZMB, NKG7 | 26 |
| FCGR3A+ Monocytes | FCGR3A, MS4A7 | 15 |
| Dendritic cells | FCER1A, CST3 | 18, 24 |
| Megakaryocytes | PPBP | 31 |
| Naive T cells | CCR7 | 2, 7, 21 |
| CD8 T cells | CD8A, CD8B | 3 |
| CD4+ T cells | IL7R, CD4 | 5, 10, 14, 35 |
| Cytotoxic T cells | NKG7, GZMA, GZMH, GZMK | 6, 11, 16, 17, 20, 25, 27, 32 |
Expression plots
raster_width <- 8 / 3
raster_height <- 8 / 4
raster_dpi <- 150
long_type_names <- c("CD14+ Monocytes", "FCGR3A+ Monocytes", "Dendritic cells",
"Naive T cells", "Cytotoxic T cells", "CD4+ T cells")
for (type in long_type_names) {
clusters_annotated[clusters_annotated == type] <- sub(" ", "\n", type)
}
gg_annotation <- PlotClustering(pgd, clusters_annotated, lineheight=0.7, size=0.2,
raster=T, raster.width=raster_width,
raster.height=raster_height, raster.dpi=raster_dpi) +
theme_pdf(show.ticks=F) + ggpubr::rremove("xylab") +
scale_size_continuous(range=c(3, 3)) +
theme(plot.margin=margin())plot_mtx <- apply(log_mtx, 2, function(vec) scales::rescale(rank(vec)))plot_genes <- c('MS4A1', 'LYZ',
'FCGR3A', 'FCER1A', 'PPBP',
'CD3D', 'CCR7', 'CD8A', 'IL7R',
'NKG7', 'GZMB')
gene_plots <- lapply(plot_genes, PlotGeneFraction, pgd, plot_mtx, title.x=0.04,
title.y=0.99, legend.position="none", size=0.2,
raster=T, raster.width=raster_width,
raster.height=raster_height, raster.dpi=raster_dpi)
gene_plots <- c(list(gg_annotation), gene_plots)
gg_fig <- cowplot::plot_grid(plotlist=gene_plots, ncol=3) +
theme(plot.margin=margin(1, 1, 1, 1))gg_fig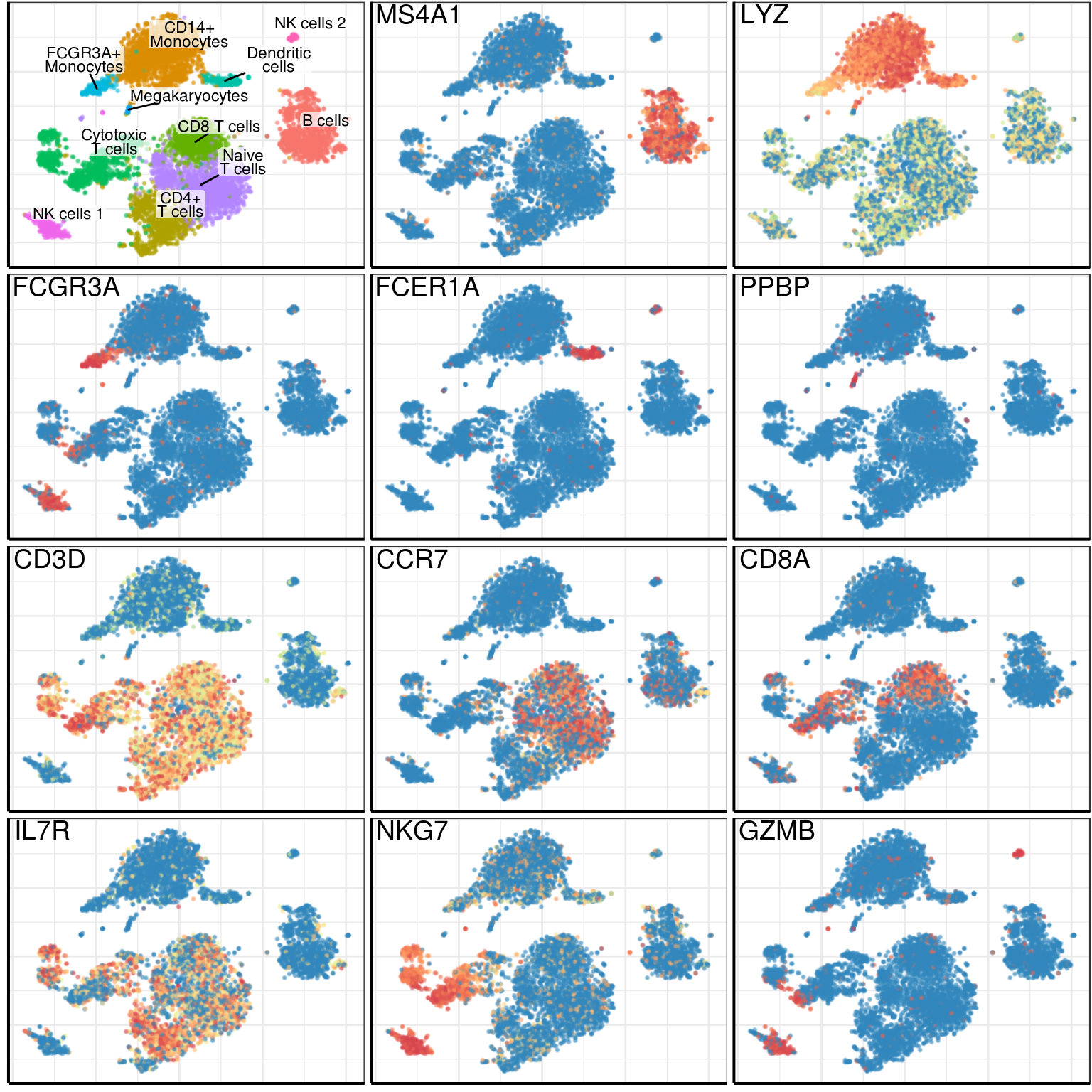
ggsave(paste0(kOutputPath, 'figures/supp_annotation_pbmc8k.pdf'), width=8, height=8)Session information
data.frame(value=unlist(sessioninfo::platform_info()))| value | |
|---|---|
| version | R version 3.4.1 (2017-06-30) |
| os | Ubuntu 14.04.5 LTS |
| system | x86_64, linux-gnu |
| ui | X11 |
| language | (EN) |
| collate | en_US.UTF-8 |
| tz | America/New_York |
| date | 2018-05-29 |
as.data.frame(sessioninfo::package_info())[c('package', 'loadedversion', 'date', 'source')]| package | loadedversion | date | source | |
|---|---|---|---|---|
| 1 | AnnotationDbi | 1.32.3 | 2016-01-28 | Bioconductor |
| 2 | assertthat | 0.2.0 | 2017-04-11 | CRAN (R 3.4.1) |
| 3 | backports | 1.1.2 | 2017-12-13 | CRAN (R 3.4.1) |
| 5 | base64enc | 0.1-3 | 2015-07-28 | cran (@0.1-3) |
| 6 | bindr | 0.1 | 2016-11-13 | CRAN (R 3.4.1) |
| 7 | bindrcpp | 0.2 | 2017-06-17 | CRAN (R 3.4.1) |
| 8 | Biobase | 2.30.0 | 2016-01-28 | Bioconductor |
| 9 | BiocGenerics | 0.16.1 | 2016-01-28 | Bioconductor |
| 10 | bit | 1.1-12 | 2014-04-09 | CRAN (R 3.4.1) |
| 11 | bit64 | 0.9-7 | 2017-05-08 | CRAN (R 3.4.1) |
| 12 | blob | 1.1.0 | 2017-06-17 | CRAN (R 3.4.1) |
| 13 | brew | 1.0-6 | 2011-04-13 | CRAN (R 3.4.1) |
| 14 | Cairo | 1.5-9 | 2015-09-26 | CRAN (R 3.4.1) |
| 15 | clisymbols | 1.2.0 | 2017-05-21 | CRAN (R 3.4.1) |
| 16 | colorspace | 1.3-2 | 2016-12-14 | CRAN (R 3.4.1) |
| 18 | cowplot | 0.9.2 | 2017-12-17 | CRAN (R 3.4.1) |
| 20 | DBI | 1.0.0 | 2018-05-02 | CRAN (R 3.4.1) |
| 21 | dendsort | 0.3.3 | 2015-12-14 | cran (@0.3.3) |
| 22 | digest | 0.6.15 | 2018-01-28 | cran (@0.6.15) |
| 23 | dplyr | 0.7.4 | 2017-09-28 | CRAN (R 3.4.1) |
| 24 | dropEstAnalysis | 0.6.0 | 2018-05-16 | local (VPetukhov/dropEstAnalysis@NA) |
| 25 | dropestr | 0.7.7 | 2018-03-17 | local (@0.7.7) |
| 26 | evaluate | 0.10.1 | 2017-06-24 | CRAN (R 3.4.1) |
| 27 | ggplot2 | 2.2.1 | 2016-12-30 | CRAN (R 3.4.1) |
| 28 | ggpubr | 0.1.6 | 2017-11-14 | CRAN (R 3.4.1) |
| 29 | ggrastr | 0.1.5 | 2017-12-28 | Github (VPetukhov/ggrastr@cc56b45) |
| 30 | ggrepel | 0.7.0 | 2017-09-29 | CRAN (R 3.4.1) |
| 31 | git2r | 0.21.0 | 2018-01-04 | cran (@0.21.0) |
| 32 | glue | 1.2.0 | 2017-10-29 | CRAN (R 3.4.1) |
| 33 | GO.db | 3.2.2 | 2017-11-12 | Bioconductor |
| 37 | gtable | 0.2.0 | 2016-02-26 | CRAN (R 3.4.1) |
| 38 | highr | 0.6 | 2016-05-09 | CRAN (R 3.4.1) |
| 39 | htmltools | 0.3.6 | 2017-04-28 | CRAN (R 3.4.1) |
| 40 | igraph | 1.2.1 | 2018-03-10 | cran (@1.2.1) |
| 41 | IRanges | 2.4.8 | 2016-09-15 | Bioconductor |
| 42 | irlba | 2.3.2 | 2018-01-11 | cran (@2.3.2) |
| 43 | KernSmooth | 2.23-15 | 2015-06-29 | CRAN (R 3.4.0) |
| 44 | knitr | 1.20 | 2018-02-20 | cran (@1.20) |
| 45 | ks | 1.11.0 | 2018-01-16 | local (VPetukhov/ks@NA) |
| 46 | labeling | 0.3 | 2014-08-23 | CRAN (R 3.4.1) |
| 47 | lattice | 0.20-35 | 2017-03-25 | CRAN (R 3.4.1) |
| 48 | lazyeval | 0.2.1 | 2017-10-29 | CRAN (R 3.4.1) |
| 49 | magrittr | 1.5 | 2014-11-22 | CRAN (R 3.4.1) |
| 50 | MASS | 7.3-47 | 2017-04-21 | CRAN (R 3.4.0) |
| 51 | Matrix | 1.2-12 | 2017-11-16 | CRAN (R 3.4.1) |
| 52 | mclust | 5.4 | 2017-11-22 | CRAN (R 3.4.1) |
| 53 | memoise | 1.1.0 | 2017-04-21 | CRAN (R 3.4.1) |
| 55 | mgcv | 1.8-22 | 2017-09-19 | CRAN (R 3.4.1) |
| 56 | munsell | 0.4.3 | 2016-02-13 | CRAN (R 3.4.1) |
| 57 | mvtnorm | 1.0-7 | 2018-01-26 | cran (@1.0-7) |
| 58 | nlme | 3.1-131 | 2017-02-06 | CRAN (R 3.4.0) |
| 59 | pagoda2 | 0.0.0.9002 | 2018-04-08 | local (hms-dbmi/pagoda2@NA) |
| 61 | pcaMethods | 1.60.0 | 2017-11-12 | Bioconductor |
| 62 | pcaPP | 1.9-73 | 2018-01-14 | cran (@1.9-73) |
| 63 | pheatmap | 1.0.8 | 2015-12-11 | CRAN (R 3.4.1) |
| 64 | pkgconfig | 2.0.1 | 2017-03-21 | CRAN (R 3.4.1) |
| 65 | plyr | 1.8.4 | 2016-06-08 | CRAN (R 3.4.1) |
| 66 | R6 | 2.2.2 | 2017-06-17 | CRAN (R 3.4.1) |
| 67 | RColorBrewer | 1.1-2 | 2014-12-07 | CRAN (R 3.4.1) |
| 68 | Rcpp | 0.12.17 | 2018-05-18 | cran (@0.12.17) |
| 69 | rjson | 0.2.19 | 2018-05-18 | cran (@0.2.19) |
| 70 | rlang | 0.1.4 | 2017-11-05 | CRAN (R 3.4.1) |
| 71 | rmarkdown | 1.9 | 2018-03-01 | CRAN (R 3.4.1) |
| 72 | Rook | 1.1-1 | 2014-10-20 | CRAN (R 3.4.1) |
| 73 | rprojroot | 1.3-2 | 2018-01-03 | cran (@1.3-2) |
| 74 | RSQLite | 2.0 | 2017-06-19 | CRAN (R 3.4.1) |
| 75 | Rtsne | 0.14 | 2017-11-12 | Github (jkrijthe/Rtsne@1d0f926) |
| 76 | S4Vectors | 0.8.11 | 2016-01-30 | Bioconductor |
| 77 | scales | 0.5.0 | 2017-08-24 | CRAN (R 3.4.1) |
| 78 | sessioninfo | 1.0.0 | 2017-06-21 | CRAN (R 3.4.1) |
| 81 | stringi | 1.1.7 | 2018-03-12 | cran (@1.1.7) |
| 82 | stringr | 1.3.0 | 2018-02-19 | cran (@1.3.0) |
| 83 | tibble | 1.3.4 | 2017-08-22 | CRAN (R 3.4.1) |
| 85 | triebeard | 0.3.0 | 2016-08-04 | cran (@0.3.0) |
| 86 | urltools | 1.7.0 | 2018-01-20 | cran (@1.7.0) |
| 88 | withr | 2.1.2 | 2018-03-15 | cran (@2.1.2) |
| 89 | yaml | 2.1.18 | 2018-03-08 | cran (@2.1.18) |
This R Markdown site was created with workflowr